Preparing some genomic annotations
Jun 3 2016 genomeMappability track
I produced a mappability track from the UCSC track. The raw file contains, for each base in the genome, an estimation of the probability that a read is correctly mapped at this position.
Using a sliding-window approach, I compute the average mappability in regions of size 1 Kbp. This is a more manageable amount of data and still informative, especially when interested in large regions (e.g. SVs).
I used a custom Perl script to efficiently parse the bedGraph-transformed original file. See the code on GitHub.
I uploaded the result there: https://dl.dropboxusercontent.com/s/i537zjs65dpw34n/map100mer-1kbp.bed.gz?dl=0.
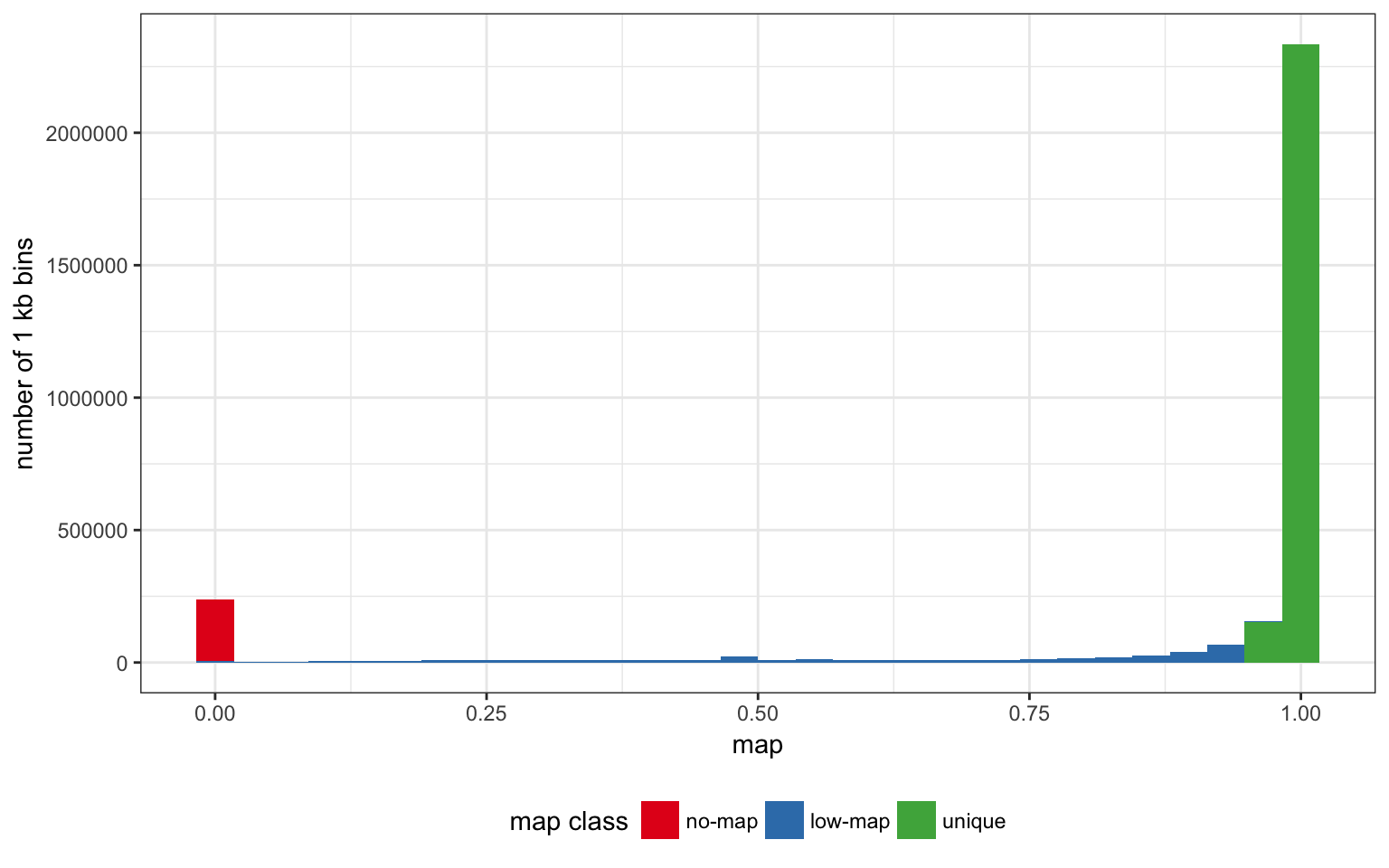
We can cut the genome into three mappability classes:
- unique regions with high mappability estimate (>0.95).
- low-map regions with a non-null mappability but lower than 0.95.
- no-map regions with mappability 0.
| map.class | Mb | prop |
|---|---|---|
| unique | 2485.972 | 0.803 |
| low-map | 375.608 | 0.121 |
| no-map | 233.228 | 0.075 |